It is possible to visualize certain cell organelles and structures that are invisible
with bright-field (transparent objects to be seen by using the difference in light’s
refraction)
The specimen will appear bright in contrast to the dark background
Able to use a full width condenser aperture setting resulting in a brighter image
No halo effect occurs with differential interference contrast
Gives a greater depth of focus - can produce very clear images of thick specimens
Can be used in conjunction with fluorescence microscopy
Suitable for living cells (long time lapses can be acquired
The three-dimensional image of a specimen may not be accurate.
The enhanced areas of light and shadow might add distortion to the appearance of the
image
Widefield Fluorescence Microscopy
Allows labelling of organelles, molecules and other features of interest
Allows tracking the dynamics of processes involving labeled features in real-time
and in vivo
The technique is highly sensitive: can detect a few molecules per cubic micrometer
Location of structures too small to be visible in a light microscope
Possibility to use different colors to track distinct molecules
Multicolor fluorescence microscopy allows to address possible interactions between
molecules by observing colocalization
Quantitative imaging
Photobleaching - dyes become nonfluorescent due to its molecular structure being altered
as a result of exposure to excitation light)
Phototoxicity - cells become damaged due to interaction between fluorescent dye and
excitation light
Inability to show morphology of surrounding structures.
Chromatic and spherical aberration
The availability of target specific antibodies
Limited specificity of the antibody
Limited ability of the antibody to diffuse to the target
Confocal Microscopy
Better vertical resolution
The ability to serially produce thin optical sections through fluorescent specimens
that have a thickness ranging up to 50 micrometers or more
Better horizontal resolution
Image information is restricted to a well-defined plane, rather than being complicated
by signals arising from remote locations in the specimen
More efficient use of light (requires less intense light, minimize photodamage)
Reduction in background fluorescence
Improved signal-to-noise
Optical sectioning of both living and fixed specimens
The ability to adjust magnification electronically by varying the area scanned by
the laser without having to change objectives
Improved quantitative imaging
The limited number of excitation wavelengths available with common lasers
Harmful nature of high-intensity laser irradiation to living cells and tissues
High quality images may require significant acquisition times
Bright-field Microscopy
The optics do not change the color of the observed structures.
Stains are used to make certain structures visible.
Bright-field microscopy requires fewer adjustments before one can observe the specimens.
Can be used to view fixed specimens or live cells.
Frees fluorescent channels in microscopes
Eliminates distortions caused by the overlapping of the color emissions of the stains
and the excitation of the fluorescing materials.
There are relatively cheap, fast and simple staining protocols to visualize:
The nuclei and cytoplasm (Haematoxylin and Eosin Staining, Methylene Blue
Neutral/Toluylene Red, Nile Blue)
Types of cells (Papanicolaou staining)
Cell walls (Crystal Violet with Mordant)
Bacteria (Giemsa stain, Gimenez stain)
Spores (Malachite Green)
Intracellular lipid globules (Nile Red)
Lipids (Osmium Tetroxide)
Collagen (Fuchsin, Safranin)
Starch (Iodine)
Mitochondria (Fuchsin)
Proteins (Coomassie Blue)
Glycogen (Carmine)
Mucins (Bismarck Brown)
low contrast
most cells must be stained to be seen
staining may introduce extraneous details
intense light used for high magnification applications can damage specimens or kill
living cells
Time Lapse Imaging
Time-lapse imaging (serial images taken at regular time points to capture the dynamics
process) can be performed using phase contrast, DIC, fluorescence, and confocal microcopy
modes.
Image Analysis
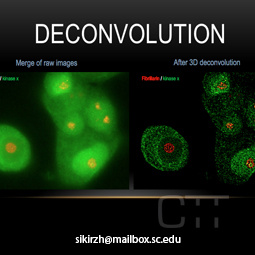
Deconvolution
A computationally intensive image processing technique which improves the contrast
and resolution of digital images captured in the microscope.
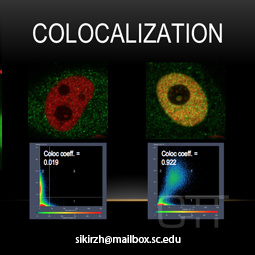
Colocalization
Colocalization determines the exact location of cellular structures of interest, and
allows for features that they have in common to be examined quantitatively.
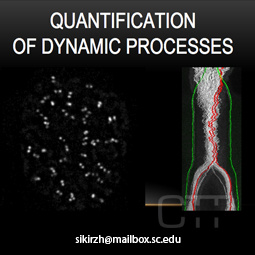
Quantification of Dynamic Processes
Application of advanced bioimaging techniques to extract quantitative data from dynamic
processes.
Challenge the conventional. Create the exceptional. No Limits.